Sample-basierte Quantediagonalisieerung von'm Chemie-Hamiltonian
Laufzeitleschätzung: unnerm Minuddn uff'm Heron-r2-Brozässor (HINWEIS: Des is bloß 'ne Schätzung. Dei Laufzeit gann abweichn.)
Hintergrund
In däm Tutorial zachn mer, wie mer verrauschte Quantesamples noochbarbeite gann, um 'ne Annäherung an'n Grundsuschstand vom Stickstoffmolekül in der Glaachgewichts-Bindungslänge ze finden — mit'm Sample-basieerdn Quantediagonalisieerungs-Algorithmus (SQD), fier Samples aus'm 59-Qubit-Quanteschaltgrees (52 Siestemqubits + 7 Ancilla-Qubits). 'ne Qiskit-basierte Implementierung is in de SQD-Qiskit-Addons verfügbar; mehr Details fiendsde in de äntschbrechendn Dokus mit'm eenfachn Beispiel zum Einschdeeche.
SQD is 'ne Technik, um Eigenwärde un Eigenvegtorn von Quanteobberatorien ze finden — wie zum Beispiel'n Hamiltonian von'm Quantesiesteem —, indem Quante- un värteiltes glassisches Rechn zusaamm eingsetzt wärn. Glassisches värteildes Rechn wird benutzt, um Samples von'm Quantebrozässor ze verarbeeden un'n Ziell-Hamiltonian in'm von ihn aufgespanndn Unterraumm ze brojiziern un diagonalisiern. Dodruch is SQD robust geegnüber verrauschdn Samples un gann mit großn Hamiltonians umgehn — wie Chemiehamiltonians mit Millionen von Wechselwirkungsgliedern —, wo jede exagte Diagonalisieerungsmethood scheitert. En SQD-basierter Workflow hot die folgenden Schridde:
- Wähl'n Schaltgrees-Ansatz un wend ihn uff'm Quantecomputer uff'n Referenzsuschstand oo (in däm Fall dr Hartree-Fock-Suschstand).
- Sample Bitfolchn von dem ergebnisweschn Quantesuschstand.
- Fiehr de Prozedur zur selbstkonsistenten Konfigurationswiederherstellung uff de Bitfolchn durch, um die Annäherung an'n Grundsuschstand ze ärhaldn.
SQD funktscheneert gut, wenn dr Zieleigensuschstand dünn besetzt is: Die Wellenfunktschn is in'm Satz von Basissuschständn untergedrächt, dessen Größe nich exbonenziell mit der Problemgröße wächst.
Quantechemie
Die Eigenschoftn von Molegüln wärn großddeels druch de Struktur von de Ellektronen in ihn bestimmt. Als fermionische Deilchen gönnen Ellektronen mit'm mathemadschen Formalismus beschribn wärn, den mer als zweide Quantisierung bezeichnet. Die Idee: Es gibt 'ne Ooziehe von Orbitalen, von denen jedes entweder leer oder von'm Fermion besetzt sei gann. En Siestem von Orbitalen wärd durch'n Satz von fermionischn Vernichtungsopperatorien beschribn, die de fermionischn Antikommutatschensrelatschen ärsatten:
Dr Adjungierte wärd als Erzeugungsoppgerator bezeichnet.
Bisher hot unsere Darschtelling dr Spin noch nich berücksichtiegt, dr 'ne grundlächende Eigenschaft von Fermionen is. Wenn mer dr Spin berücksichtiegt, kommen de Orbitale in Paarchen, die mer als Raumorbidaale bezeichnet. Jedes Raumorbital besteht aus zwei Spinorbitalen, einem mit der Beziechnung „Spin-" un einm mit der Beziechnung „Spin-". Mer schreiben dann fier dr Vernichtungsoppgerator, der zum Spinorbital mit'm Spin () im Raumorbital ghört. Wenn mer als die Ooziehe von Raumorbidaalen nähmt, dann gibts insgesamt Spinorbitale. Dr Hilbertraum von däm Siestem wird von ordonormalen Basisvegtoern aufgespannt, die mit zweideilichen Bitfolchn bezeichnet wärn.
Dr Hamiltonian von'm Molegülsiestem gann geschriebn wärn als
wobei die un komplexe Zahlen sinn, die mer als Molegülintegrale bezeichnet un die aus der Spezifigatschn vom Molekül mit'm Computerprogramm bereechnet wärn könn. In däm Tutorial bereechnen mer die Integrale mit'm Softwarepaket PySCF.
Fier Details darüber, wie der molegülare Hamiltonian hergeleitet wärd, schau'n de Lehrbücher zur Quantechemie oo (zum Beispiel Modern Quantum Chemistry von Szabo un Ostlund). Fier'ne übberschlächschde Ärglärung, wie Quantechemieprobleme uff Quantecomputer abgebildet wärn, guggsde die Vorlesung Mapping Problems to Qubits vom Qiskit Global Summer School 2024 oo.
Lokaler Unitärer Cluster-Jastrow-Ansatz (LUCJ)
SQD brauchd'n Schaltgrees-Ansatz, aus dem Samples gezochn wärn. In däm Tutorial nutzn mer'n lokalen unitärn Cluster-Jastrow-Ansatz (LUCJ) wechn sei'r Kombination aus fieesikalischer Motivatschn un Hardwarefreundlichkeet.
Dr LUCJ-Ansatz is 'ne spezialisierte Form vom allgemein unitärn Cluster-Jastrow-Ansatz (UCJ), dr die Form hot
wobei 'n Referenzsuschstand is — oft'n Hartree-Fock-Suschstand —, un de un die Form habn:
wobei mer'n Zoaloppgerator definiert habn:
Dr Oppgerator is 'ne Orbitalrotatschn, die mit bekannten Algorithmen in linearer Diefe un mit linearer Konnektivität implementiert wärn gann. Dr -Term vom UCJ-Ansatz ze implementiern erfordert entweder allgemeine Konnektivität oder de Nutzung von'm Fermion-Swap-Netzwerk, was fier verrauschde prä-fählertolerante Quantebrozässorn mit begrenzter Konnektivität schwierig is. Die Idee vom lokalen UCJ-Ansatz is es, Dünnheitseinschränkungen an de - un -Matritzen aufzuerlegen, die ihn in gonschdanter Diefe uff Qubit-Topologien mit begrenzter Konnektivität implementierbar machn. Die Einschränkungen wärn druch 'ne Liste von Indizes angegäbn, de anzeichn, welche Matritzeneinträge im obern Dreieck von Null verschieden sein dürfn (da die Matritzen siemmetrisch sinn, muss nur 's obere Dreieck angegäbn wärn). Die Indizes gönnen als Orbitalpaare interprediiert wärn, die miteinander wechselwirken dürfn.
Als Beispiel betrachte 'ne quadratische Gidder-Qubit-Topologie. Mer gann de - un -Orbitale in paralleln Linien uffm Gidder platziern, wobei Verbindungen zwischn diesen Linien „Sprossen" von'r Leiternform bilden, so wie hier:

Mit däm Aufbau sinn Orbitale mit gleichm Spin mit'r Linientopologie verbunden, während Orbitale mit verschiedenem Spin verbunden sinn, wenn se das gleiche Raumorbital deeln. Des ergitt die folgenden Indexeinschränkungen fier de -Matritzen:
Mit andern Worten: Wenn de -Matritzen nur oo de angegebnen Indizes im obern Dreieck von Null verschieden sinn, dann gann dr -Term uff'r quadratischn Topologie ohne Swap-Gates in gonschdanter Diefe implementiert wärn. Natierlich macht des dr Ansatz weniger ausdrucksstarg, sodass mehr Ansatz-Wiederhoolungen nöddsch sein gönnen.
Die IBM®-Hardware hot 'ne Heavy-Hex-Gidder-Qubit-Topologie; in däm Fall gann mer 'n „Zickzack"-Muster oobäwendn, wie unnern dargestellt. In däm Muster wärn Orbitale mit gleichm Spin uff Qubits mit Linientopologie abgebildet (rode un blaue Kreese), un 'ne Verbindung zwischn Orbitalen mit verschiedenem Spin is bei jedem 4. Raumorbital vorhanden, wobei die Verbindung druch'n Ancilla-Qubit (lila Kreese) ärmöchlicht wärd. In däm Fall sinn die Indexeinschränkungen:
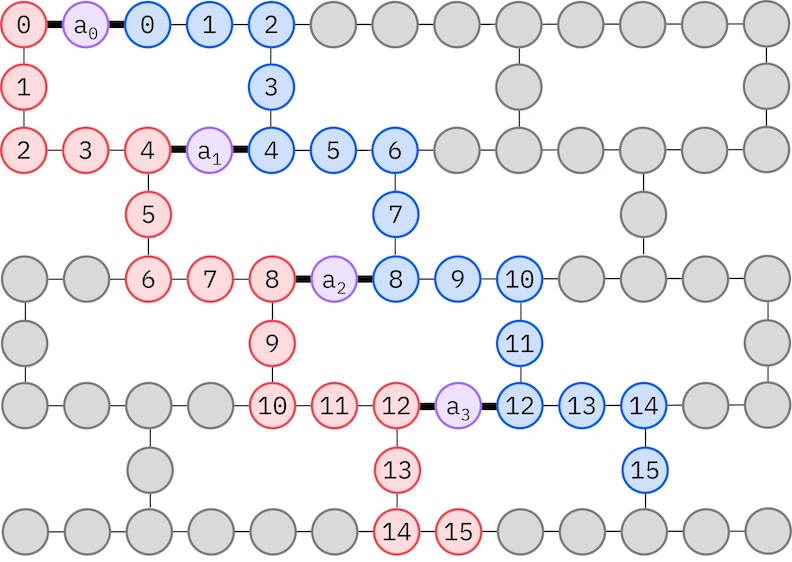
Selbstkonsistente Konfigurationswiederherstellung
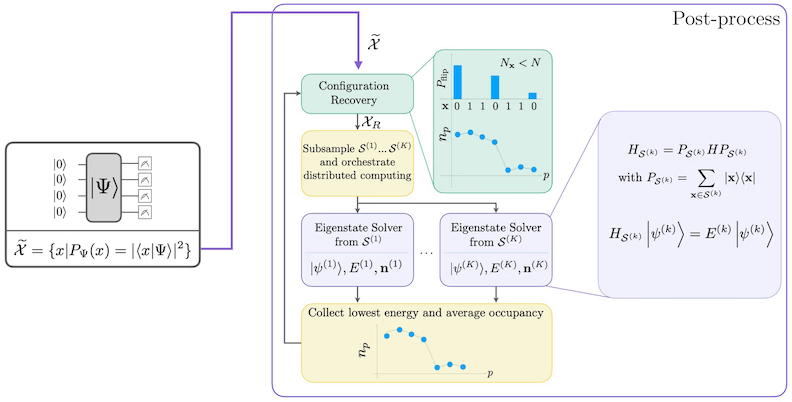
Die Prozedur zur selbstkonsistenten Konfigurationswiederherstellung is dafier gedacht, so viel Signal wie mächlisch aus verrauschdn Quantesamples ze extrahiern. Weil dr molegülare Hamiltonian die Deilchenzaal un dr Spin-Z erhält, is es sinnvoll,'n Schaltgrees-Ansatz ze wähln, der diese Siemmetrien ebenfalls behält. Wenns uff'n Hartree-Fock-Suschstand oongwandt wärd, hot dr ergebnisweschliche Suschstand ohn Rauschen 'ne feste Deilchenzaal un Spin-Z. Deshalb solln de Spin-- un Spin--Hälften jeder aus däm Suschstand gezognen Bitfolch das gleiche Hamming-Gewicht wie im Hartree-Fock-Suschstand habn. Druch dr Präsenz von Rauschen in gägenweärschn Quantebrozässorn wärn manche gemässnen Bitfolchn diese Eigenschaft verletzn. 'ne eenfache Nachauswaal würde diese Bitfolchn verwerfn, aber des is verschwendungsreich, weil die Bitfolchn trotzdem noch Signal enthaldn könntn. Die selbstkonsistente Wiederherstellungsprozedur versucht, enich von däm Signal in der Noochbarbeitung zurückzegewinnen. Die Prozedur is iterativ un brauchd als Eingabe 'ne Schätzung von de drchschnittlichen Besetzungszoaln jedes Orbitals im Grundsuschstand, die zunächst aus de rohen Samples bereechnet wärd. Die Prozedur wärd in'r Schleife ausgefiehrt, un jede Iteration hot die folgenden Schridde:
- Fier jede Bitfolch, die die angegebnen Siemmetrien verletzt, kippe de Bits mit'r stochastischen Prozedur, die darauf ausgerichtet is, die Bitfolch näher oo die gägenwertige Schätzung von de drchschnittlichen Orbitalbesetzungen ze bringen, um 'ne neue Bitfolch ze ärhaldn.
- Sammle alle aldn un neuen Bitfolchn, die de Siemmetrien erfülln, un samplere Untermengen von'r festgelächdn Größe, die vorher bestimmt wordn sinn.
- Fier jede Untermenge von Bitfolchn, brojizier'n Hamiltonian in'n von de äntschbrechendn Basisvegtorn aufgespanndn Unterraum (sieh dr vorigschn Abschnitt fier 'ne Beschreibung von diesen Basisvegtorn), un bereechne uff'm glassischn Computer 'ne Grundsuschstandsschätzung vom brojizieertn Hamiltonian.
- Aktualisiere die Schätzung von de drchschnittlichen Orbitalbesetzungen mit der Grundsuschstandsschätzung mit der niedrigschddn Energie.
SQD-Workflow-Diagramm
Dr SQD-Workflow wärd im folgenden Diagramm dargestellt:
Voraussetzngen
Vor'm Starten von däm Tutorial stell siech'r, dass folgendes installiert is:
- Qiskit SDK v1.0 oder neeer, mit Untersditzung fier Visualisieerung
- Qiskit Runtime v0.22 oder neeer (
pip install qiskit-ibm-runtime) - SQD-Qiskit-Addon v0.11 oder neeer (
pip install qiskit-addon-sqd) - ffsim v0.0.58 oder neeer (
pip install ffsim)
Einrichtung
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime rustworkx
import pyscf
import pyscf.cc
import pyscf.mcscf
import ffsim
import numpy as np
import matplotlib.pyplot as plt
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Schritt 1: Glassische Eingabn uff's Quanteproblem abbildn
In däm Tutorial fiendn mer 'ne Annäherung oo'n Grundsuschstand vom Molekül in der Glaachgewichtslage im cc-pVDZ-Basissatz. Zunächst lächn mer 's Molekül un sei Eigenschoftn fest.
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Store reference energy from SCI calculation performed separately
exact_energy = -109.22690201485733
converged SCF energy = -108.929838385609
Bevor mer'n LUCJ-Ansatz-Schaltgrees konstruiern, fiehren mer im folgenden Code-Block zunächst 'ne CCSD-Bereechnung durch. Die - un -Amplituden aus däser Bereechnung wärn zur Initialisierung von de Parametern vom Ansatz genutzt.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.2177884185543 E_corr = -0.2879500329450045
Jetz nutzen mer ffsim, um'n Ansatz-Schaltgrees ze erschaffa. Da unser Molekül'n geschlossenschaaligen Hartree-Fock-Suschstand hot, nutzn mer die spinausglaachene Variante vom UCJ-Ansatz, UCJOpSpinBalanced. Mer übergäbn Wechselwirkungspaare, die fier 'ne Heavy-Hex-Gidder-Qubit-Topologie geeignet sinn (sieh'n Hintergrundabschnitt zum LUCJ-Ansatz). Mer setzn optimize=True in der from_t_amplitudes-Methode, um die „komprimierte" Doppelfaktorisieerung von de -Amplituden zu aktiviern (sieh de ffsim-Dokus zum Thema The local unitary cluster Jastrow (LUCJ) ansatz fier Details).
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell from running
# too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Schritt 2: Problem fier die Quantehardware-Ausfiehrung optimiern
Als Nächstes optimiern mer'n Schaltgrees fier 'ne Zielhardware.
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
Using backend ibm_fez
Mer ämbfehln die folgenden Schridde, um'n Ansatz ze optimiern un mit der Hardware kompatibel ze machn.
- Wähl fieesikalische Qubits (
initial_layout) von der Zielhardware aus, die'm vor'r beschriebnen „Zickzack"-Muster äntschbrechn. De Qubits in däm Muster oozeordnen fiehrt ze'm effizienten hardwarekompatibeln Schaltgrees mit weniger Gates. Hier gibts Code, der die Auswaal vom „Zickzack"-Musters automatisiert und 'ne Bewertungsheuristik einsetzt, um die Fehler von der gewähltn Ooordnung ze minimiern. - Generier'n gestufte Pass-Manager mit der generate_preset_pass_manager-Funktion aus Qiskit mit dein'm gewähltn
backenduninitial_layout. - Setz de
pre_init-Stuf von dein'm gestuftn Pass-Manager uffffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITenthält Qiskit-Transpiler-Pässe, die Gates in Orbitalrotatschenen zerlegn un die Orbitalrotatschenen dann zusammenfiehrn, was zu weniger Gates im abschließenden Schaltgrees führt. - Fiehr'n Pass-Manager uff deim Schaltgrees aus.
Code fier automatisierte Auswaal vom „Zickzack"-Layout
from typing import Sequence
import rustworkx
from qiskit.providers import BackendV2
from rustworkx import NoEdgeBetweenNodes, PyGraph
IBM_TWO_Q_GATES = {"cx", "ecr", "cz"}
def create_linear_chains(num_orbitals: int) -> PyGraph:
"""In zig-zag layout, there are two linear chains (with connecting qubits between
the chains). This function creates those two linear chains: a rustworkx PyGraph
with two disconnected linear chains. Each chain contains `num_orbitals` number
of nodes, that is, in the final graph there are `2 * num_orbitals` number of nodes.
Args:
num_orbitals (int): Number orbitals or nodes in each linear chain. They are
also known as alpha-alpha interaction qubits.
Returns:
A rustworkx.PyGraph with two disconnected linear chains each with `num_orbitals`
number of nodes.
"""
G = rustworkx.PyGraph()
for n in range(num_orbitals):
G.add_node(n)
for n in range(num_orbitals - 1):
G.add_edge(n, n + 1, None)
for n in range(num_orbitals, 2 * num_orbitals):
G.add_node(n)
for n in range(num_orbitals, 2 * num_orbitals - 1):
G.add_edge(n, n + 1, None)
return G
def create_lucj_zigzag_layout(
num_orbitals: int, backend_coupling_graph: PyGraph
) -> tuple[PyGraph, int]:
"""This function creates the complete zigzag graph that 'can be mapped' to an IBM QPU with
heavy-hex connectivity (the zigzag must be an isomorphic sub-graph to the QPU/backend
coupling graph for it to be mapped).
The zigzag pattern includes both linear chains (alpha-alpha interactions) and connecting
qubits between the linear chains (alpha-beta interactions).
Args:
num_orbitals (int): Number of orbitals, that is, number of nodes in each alpha-alpha linear chain.
backend_coupling_graph (PyGraph): The coupling graph of the backend on which the LUCJ ansatz
will be mapped and run. This function takes the coupling graph as a undirected
`rustworkx.PyGraph` where there is only one 'undirected' edge between two nodes,
that is, qubits. Usually, the coupling graph of a IBM backend is directed (for example, Eagle devices
such as ibm_brisbane) or may have two edges between two nodes (for example, Heron `ibm_torino`).
A user needs to be make such graphs undirected and/or remove duplicate edges to make them
compatible with this function.
Returns:
G_new (PyGraph): The graph with IBM backend compliant zigzag pattern.
num_alpha_beta_qubits (int): Number of connecting qubits between the linear chains
in the zigzag pattern. While we want as many connecting (alpha-beta) qubits between
the linear (alpha-alpha) chains, we cannot accommodate all due to qubit and connectivity
constraints of backends. This is the maximum number of connecting qubits the zigzag pattern
can have while being backend compliant (that is, isomorphic to backend coupling graph).
"""
isomorphic = False
G = create_linear_chains(num_orbitals=num_orbitals)
num_iters = num_orbitals
while not isomorphic:
G_new = G.copy()
num_alpha_beta_qubits = 0
for n in range(num_iters):
if n % 4 == 0:
new_node = 2 * num_orbitals + num_alpha_beta_qubits
G_new.add_node(new_node)
G_new.add_edge(n, new_node, None)
G_new.add_edge(new_node, n + num_orbitals, None)
num_alpha_beta_qubits = num_alpha_beta_qubits + 1
isomorphic = rustworkx.is_subgraph_isomorphic(
backend_coupling_graph, G_new
)
num_iters -= 1
return G_new, num_alpha_beta_qubits
def lightweight_layout_error_scoring(
backend: BackendV2,
virtual_edges: Sequence[Sequence[int]],
physical_layouts: Sequence[int],
two_q_gate_name: str,
) -> list[list[list[int], float]]:
"""Lightweight and heuristic function to score isomorphic layouts. There can be many zigzag patterns,
each with different set of physical qubits, that can be mapped to a backend. Some of them may
include less noise qubits and couplings than others. This function computes a simple error score
for each such layout. It sums up 2Q gate error for all couplings in the zigzag pattern (layout) and
measurement of errors of physical qubits in the layout to compute the error score.
Note:
This lightweight scoring can be refined using concepts such as mapomatic.
Args:
backend (BackendV2): A backend.
virtual_edges (Sequence[Sequence[int]]): Edges in the device compliant zigzag pattern where
nodes are numbered from 0 to (2 * num_orbitals + num_alpha_beta_qubits).
physical_layouts (Sequence[int]): All physical layouts of the zigzag pattern that are isomorphic
to each other and to the larger backend coupling map.
two_q_gate_name (str): The name of the two-qubit gate of the backend. The name is used for fetching
two-qubit gate error from backend properties.
Returns:
scores (list): A list of lists where each sublist contains two items. First item is the layout, and
second item is a float representing error score of the layout. The layouts in the `scores` are
sorted in the ascending order of error score.
"""
props = backend.properties()
scores = []
for layout in physical_layouts:
total_2q_error = 0
for edge in virtual_edges:
physical_edge = (layout[edge[0]], layout[edge[1]])
try:
ge = props.gate_error(two_q_gate_name, physical_edge)
except Exception:
ge = props.gate_error(two_q_gate_name, physical_edge[::-1])
total_2q_error += ge
total_measurement_error = 0
for qubit in layout:
meas_error = props.readout_error(qubit)
total_measurement_error += meas_error
scores.append([layout, total_2q_error + total_measurement_error])
return sorted(scores, key=lambda x: x[1])
def _make_backend_cmap_pygraph(backend: BackendV2) -> PyGraph:
graph = backend.coupling_map.graph
if not graph.is_symmetric():
graph.make_symmetric()
backend_coupling_graph = graph.to_undirected()
edge_list = backend_coupling_graph.edge_list()
removed_edge = []
for edge in edge_list:
if set(edge) in removed_edge:
continue
try:
backend_coupling_graph.remove_edge(edge[0], edge[1])
removed_edge.append(set(edge))
except NoEdgeBetweenNodes:
pass
return backend_coupling_graph
def get_zigzag_physical_layout(
num_orbitals: int, backend: BackendV2, score_layouts: bool = True
) -> tuple[list[int], int]:
"""The main function that generates the zigzag pattern with physical qubits that can be used
as an `intial_layout` in a preset passmanager/transpiler.
Args:
num_orbitals (int): Number of orbitals.
backend (BackendV2): A backend.
score_layouts (bool): Optional. If `True`, it uses the `lightweight_layout_error_scoring`
function to score the isomorphic layouts and returns the layout with less erroneous qubits.
If `False`, returns the first isomorphic subgraph.
Returns:
A tuple of device compliant layout (list[int]) with zigzag pattern and an int representing
number of alpha-beta-interactions.
"""
backend_coupling_graph = _make_backend_cmap_pygraph(backend=backend)
G, num_alpha_beta_qubits = create_lucj_zigzag_layout(
num_orbitals=num_orbitals,
backend_coupling_graph=backend_coupling_graph,
)
isomorphic_mappings = rustworkx.vf2_mapping(
backend_coupling_graph, G, subgraph=True
)
isomorphic_mappings = list(isomorphic_mappings)
edges = list(G.edge_list())
layouts = []
for mapping in isomorphic_mappings:
initial_layout = [None] * (2 * num_orbitals + num_alpha_beta_qubits)
for key, value in mapping.items():
initial_layout[value] = key
layouts.append(initial_layout)
two_q_gate_name = IBM_TWO_Q_GATES.intersection(
backend.configuration().basis_gates
).pop()
if score_layouts:
scores = lightweight_layout_error_scoring(
backend=backend,
virtual_edges=edges,
physical_layouts=layouts,
two_q_gate_name=two_q_gate_name,
)
return scores[0][0][:-num_alpha_beta_qubits], num_alpha_beta_qubits
return layouts[0][:-num_alpha_beta_qubits], num_alpha_beta_qubits
initial_layout, _ = get_zigzag_physical_layout(num_orbitals, backend=backend)
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 12438, 'sx': 12169, 'cz': 3986, 'x': 573, 'measure': 52, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'sx': 7059, 'rz': 6962, 'cz': 1876, 'measure': 52, 'x': 35, 'barrier': 1})
Schritt 3: Ausfiehrung mit Qiskit-Primitives
Noochdem mer'n Schaltgrees fier de Hardwareausfiehrung optimiert habn, sinn mer bereit, ihn uff der Zielhardware auszefiehrn un Samples fier de Grundsuschstandsenergischätzung ze sammln. Da mer nur 'n Schaltgrees habn, nutzn mer Qiskit Runtimes Job-Ausfiehrungsmodus un fiehrn unsern Schaltgrees aus.
sampler = Sampler(mode=backend)
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
Schritt 4: Noochbearbeitung un Ergebnis im gewünschdn glassischn Format zurückgäbn
Jetz schätzen mer die Grundsuschstandsenergie vom Hamiltonian mit der Funktion diagonalize_fermionic_hamiltonian. Die Funktion fiehrt die selbstkonsistente Konfigurationswiederherstellungsprozedur druch, um die verrauschdn Quantesamples iterativ ze verfeinern un die Energischätzung ze verbässern. Mer übergäbn 'ne Callback-Funktion, damit mer die Zwischenergebnisse fier 'ne spätere Analyse speichern gönnen. Sieh de API-Dokumentatschn fier Ärglärungen von de Argumendn zu diagonalize_fermionic_hamiltonian.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
pub_result.data.meas,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=12345,
)
Iteration 1
Subsample 0
Energy: -108.97781410104506
Subspace dimension: 28561
Subsample 1
Energy: -108.97781410104506
Subspace dimension: 28561
Subsample 2
Energy: -108.97781410104506
Subspace dimension: 28561
Iteration 2
Subsample 0
Energy: -109.05150860754739
Subspace dimension: 287296
Subsample 1
Energy: -109.08152283263908
Subspace dimension: 264196
Subsample 2
Energy: -109.11636893067873
Subspace dimension: 284089
Iteration 3
Subsample 0
Energy: -109.15878555367885
Subspace dimension: 429025
Subsample 1
Energy: -109.16462831275786
Subspace dimension: 473344
Subsample 2
Energy: -109.15895026995382
Subspace dimension: 435600
Iteration 4
Subsample 0
Energy: -109.17784051223317
Subspace dimension: 622521
Subsample 1
Energy: -109.1774651326829
Subspace dimension: 657721
Subsample 2
Energy: -109.18085212360191
Subspace dimension: 617796
Iteration 5
Subsample 0
Energy: -109.18636242518915
Subspace dimension: 815409
Subsample 1
Energy: -109.18451014767518
Subspace dimension: 837225
Subsample 2
Energy: -109.18333728638888
Subspace dimension: 857476
Ergebnisse visualisiern
Dr erschde Plot zacht, dass mer nooch 'n paar Iteratschonen die Grundsuschstandsenergie uff ca. ~41 mH schätzen (die chemische Gnauichkeet wärd üblicherweise als 1 kcal/mol 1,6 mH oongegäbn). Die Energie gann dursch mehr Iteratschonen von der Konfigurationswiederherstellung oder dursch die Änährung von mehr Samples pro Batch värbaässert wärn.
Dr zweede Plot zacht die drchschnittliche Besetzung jedes Raumorbidals nooch der letzten Iteratschn. Mer siehst, dass sowohl die Spin-up- als ooch de Spin-down-Ellektronen in unsern Lösungen mit hoher Wohrscheinlichkeet die erschdn fünf Orbitale besetzn.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Tutorial-Umfroage
Bitte nimm oo de'r Umfroage, um Feedback zu däm Tutorial ze gäbn. Dei Einschätzungen helfen uns, unsere Inhalte un Benutzererfahrung ze värbaässern.